Work
Research projects
- Epigenomic analysis of Neuroblastoma
- Metabolic deregulation in cancer
- Bayesian networks of epigenetic regulation
- Computational biology toolbox
- Deconvolution of immune cell types in mixed tissues
- Redox signalling in yeast
- Computational phenotyping
- Other collaboration projects
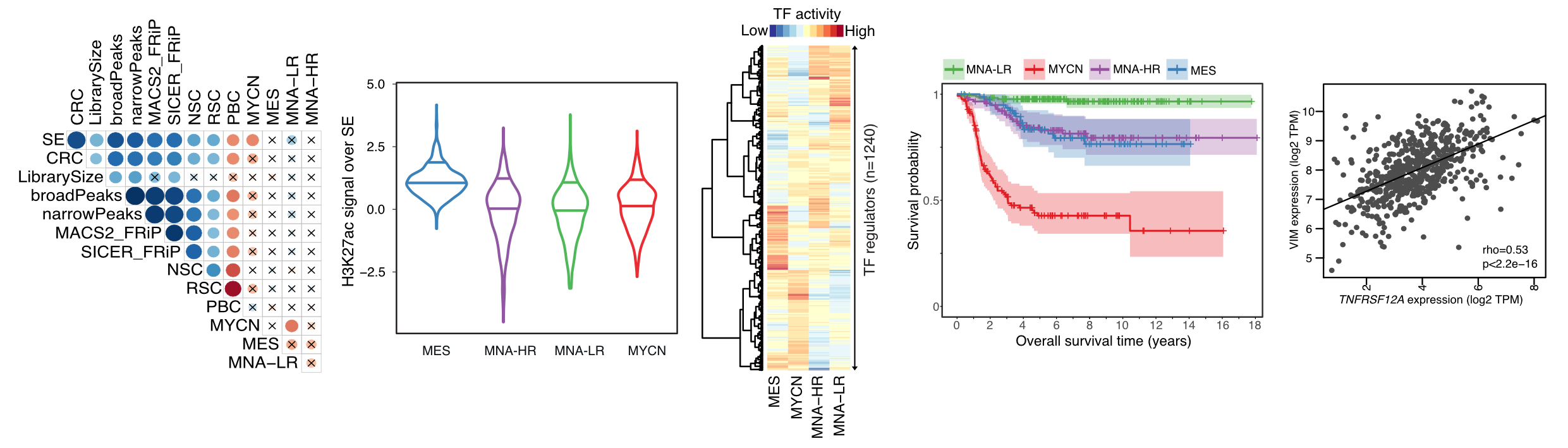
Epigenomic analysis of Neuroblastoma
Half of the children diagnosed with neuroblastoma have high risk disease, disproportionately contributing to overall childhood cancer-related deaths. In addition to recurrent gene mutations, there is increasing evidence supporting the role of epigenetic deregulation in disease pathogenesis. Yet, comprehensive cis-regulatory network descriptions from neuroblastoma are lacking. Here, using genome-wide H3K27ac profiles across 60 neuroblastomas, covering the different clinical and molecular subtypes, we identified four major super enhancer-driven epigenetic subtypes and their underlying master regulatory networks. Three of these subtypes recapitulated known clinical groups, namely MYCN-amplified, MYCN non-amplified high-risk and MYCN non-amplified low-risk neuroblastomas. The fourth subtype, exhibiting mesenchymal characteristics, shared cellular identity with multipotent Schwann cell precursors, was induced by RAS activation and was enriched in relapsed disease. Notably, CCND1, an essential gene in neuroblastoma, was regulated by both mesenchymal and adrenergic regulatory networks converging on distinct super-enhancer modules. Taken all together, this study reveals subtype-specific super enhancer regulation in neuroblastomas.
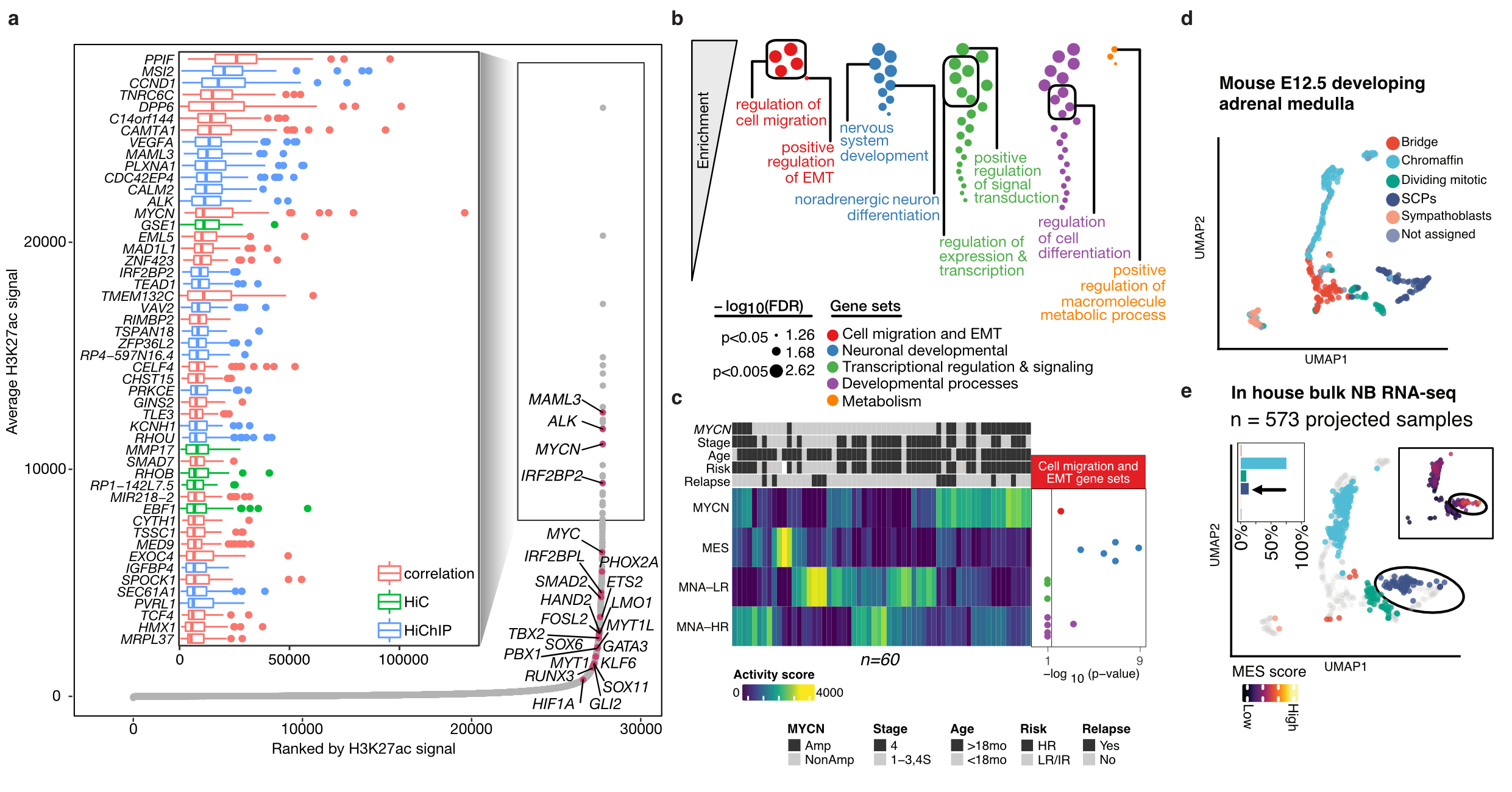
(a) Top super enhancers identified in neuroblastoma patients (b) Major biological processes regulated by these super enhancers (c) Epigenetic subtypes defined by these super enhancers are associated with clinical association (d,e) Projection of bulk tumors onto developing adrenal medulla single cell reference to identify neuroblastoma cell-of-origin
Metabolic deregulation in cancer
Somatic copy number alterations frequently occur in the cancer genome affecting not only oncogenic or tumor suppressive genes, but also passenger and potential codriver genes. An intrinsic feature resulting from such genomic perturbations is the deregulation in the metabolism of tumor cells. In this study, we have shown that metabolic and cancer-causing genes are unexpectedly often proximally positioned in the chromosome and share loci with coaltered copy numbers across multiple cancers (19 cancer types from The Cancer Genome Atlas). We have developed an analysis pipeline, Identification of Metabolic Cancer Genes (iMetCG), to infer the functional impact on metabolic remodeling from such coamplifications and codeletions and delineate genes driving cancer metabolism from those that are neutral. Using our identified metabolic genes, we were able to classify tumors based on their tissue and developmental origins. These metabolic genes were similar to known cancer genes in terms of their network connectivity, isoform frequency, and evolutionary features. We further validated these identified metabolic genes by (i) using gene essentiality data from several tumor cell lines, (ii) showing that these identified metabolic genes are strong indicators for patient survival, and (iii) observing a significant overlap between our identified metabolic genes and known cancer-metabolic genes. Our analyses revealed a hitherto unknown generic mechanism for large-scale metabolic reprogramming in cancer cells based on linear gene proximities between cancer-causing and -metabolic genes. We have identified 119 new metabolic cancer genes likely to be involved in rewiring cancer cell metabolism.
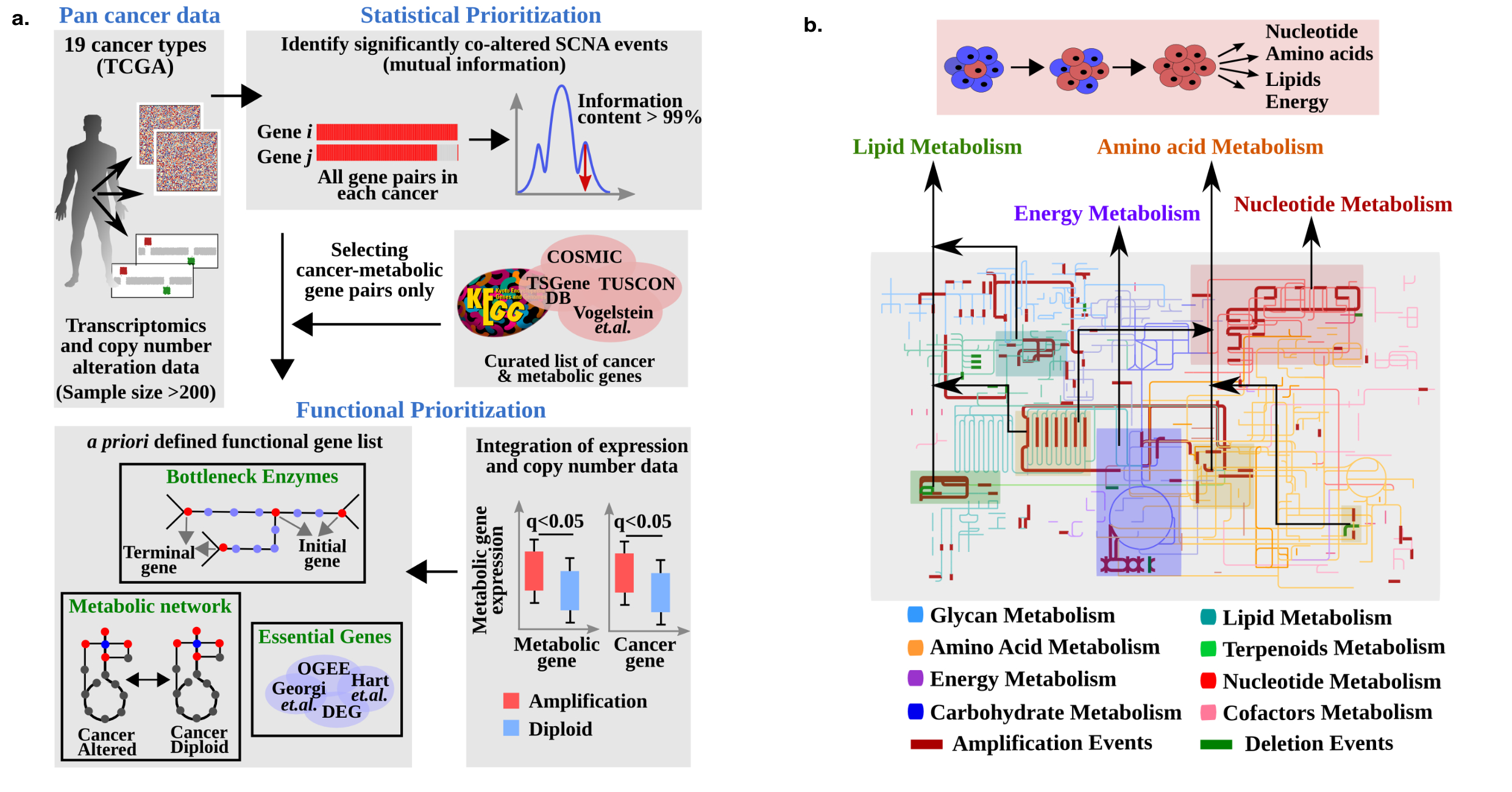
(a) Schematic representation of the iMetCG analysis pipeline. There are five steps in the iMetCG analysis for predicting metabolic cancer genes — collection of transcriptomics and copy number alterations data, statistical prioritization of all gene pairs, selection of cancer–metabolic gene pairs, correlating copy number and expression data, and integrating metabolic functionality features (b) The metabolic landscape of a normal cell is rewired to support tumorigenesis. Cancer–metabolic gene pair copy number co-alteration induced perturbations in the global human metabolic network. Representative metabolic reactions and pathways altered in cancers through copy number changes are shown.
Bayesian networks of epigenetic regulation
Gene expression is a complex molecular process regulated by a plethora of genetic and epigenetic factors acting at the proximal (promotors) and distal (enhancers) regulatory elements. While the effects of individual regulatory factors on gene expression are well elucidated, the underlying directed network structure among these regulatory features and its integrative effects on gene expression remains largely unexplored and poorly understood. In this study, we have combined various gene regulatory information at the gene level from its promotor and enhancer regions across 102 cells types covering 17 tissue types. This regulatory information pertains to 30 different histone modification states, DNA methylation, chromatin accessibility and expression. Using Bayesian structure learning, we have generated network models describing the modes of action of directed interaction among various epigenetic regulators. We observed that these network representations could discriminate between regulatory regions, differentiation states and tissue types. We also identify graph-related and epigenetic features that are most strongly responsible for resolving these differences. Next, we repeat our analysis on malignant and normal B cells from chronic lymphocytic leukaemia where we focus only on a small set of core regulatory features shown to be critical across multiple tissues in our analysis. We show that the reduced networks could not only recapitulate the differences among regulatory regions but also resolve the clinical subtypes and normal samples. These models provide a novel overview of directed epigenetic interactions present across tissues which can guide in the design of interventional epigenetic drugs in the future.
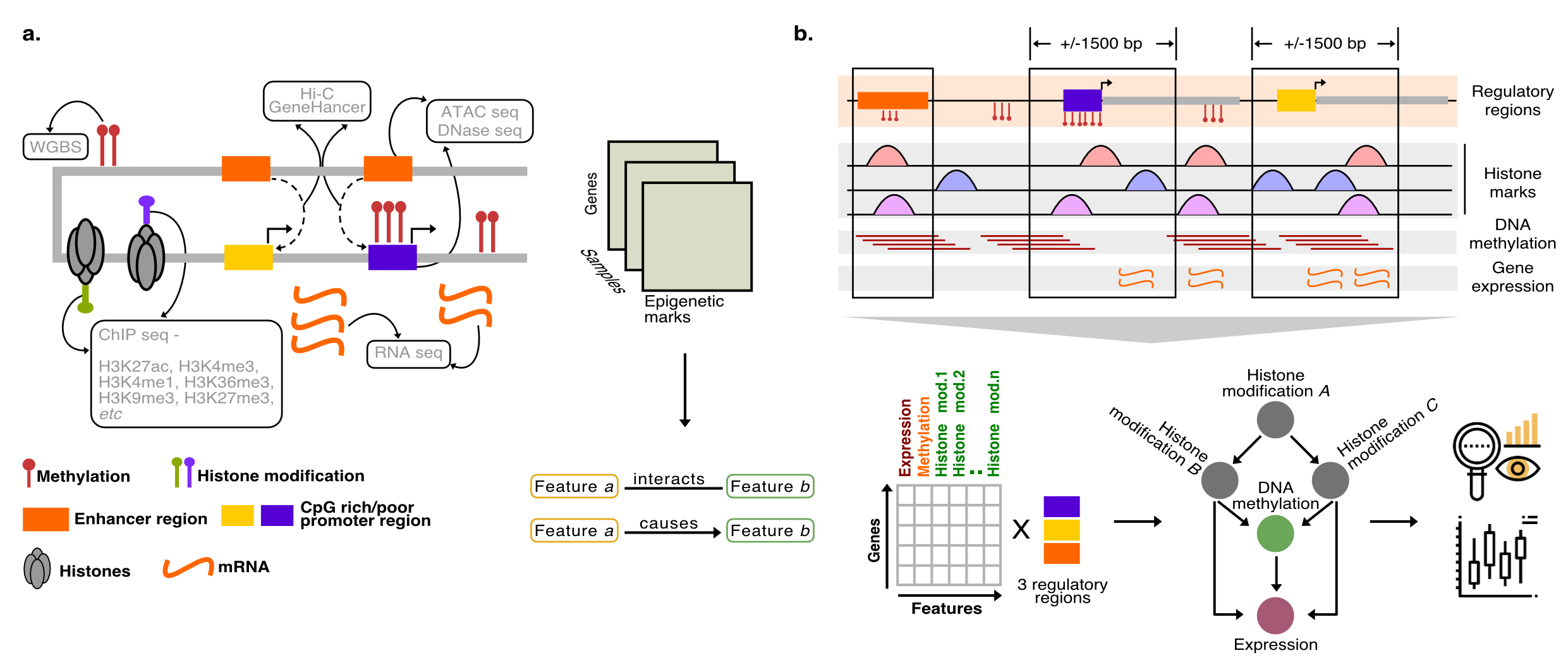
(a) Gene regulation is the product of complex interplay between cis- and trans- regulatory factors, the technologies used to quantitatively measure these factors are shown in the boxes. By integrating such information across different regulatory features across multiple tissues one could identify fundamental associations among the regulatory components (b) We want to integrate these information across multiple tissues to identify common governing principles behind transcriptional regulation
Deconvolution of immune cell types in mixed tissues
Any normal tissue (e.g Pancreas) is composed of multiple cell types (like alpha, beta, gamma etc) similarly, tumor biopsies are composed of tumor cells and its microenvironment which can consist of immune cells, fibroblasts etc. Thus, any bulk RNA sequencing data is a reflection of averaged gene expression across the constituent cell types. Computational methods have been developed to deconvolute cell type specific signals from mixed bulk signals. These methods can be supervised or unsupervised. Supervised methods rely on references/gene signatures generated from purified cells. Denovo methods on the other hand don’t need references but an expected number of cell types (k) has to be provided. Essentially all methods use regression, matrix factorization, enrichment or average expression of marker genes to accomplish this deconvolution task.
I have been involved with the COMETH project which aim to create a benchmarking platform to evaluate the accuracy of deconvolution methods. I have been involved in Dockerizing widely used Deconvolution tools to be used for benchmarking. We also aim to provide simple web based solutions where the user can upload their dataset and in return will get cell type estimates from multiple tools.
I am implementing these solutions in the de.NBI cloud based on OpenStack framework.
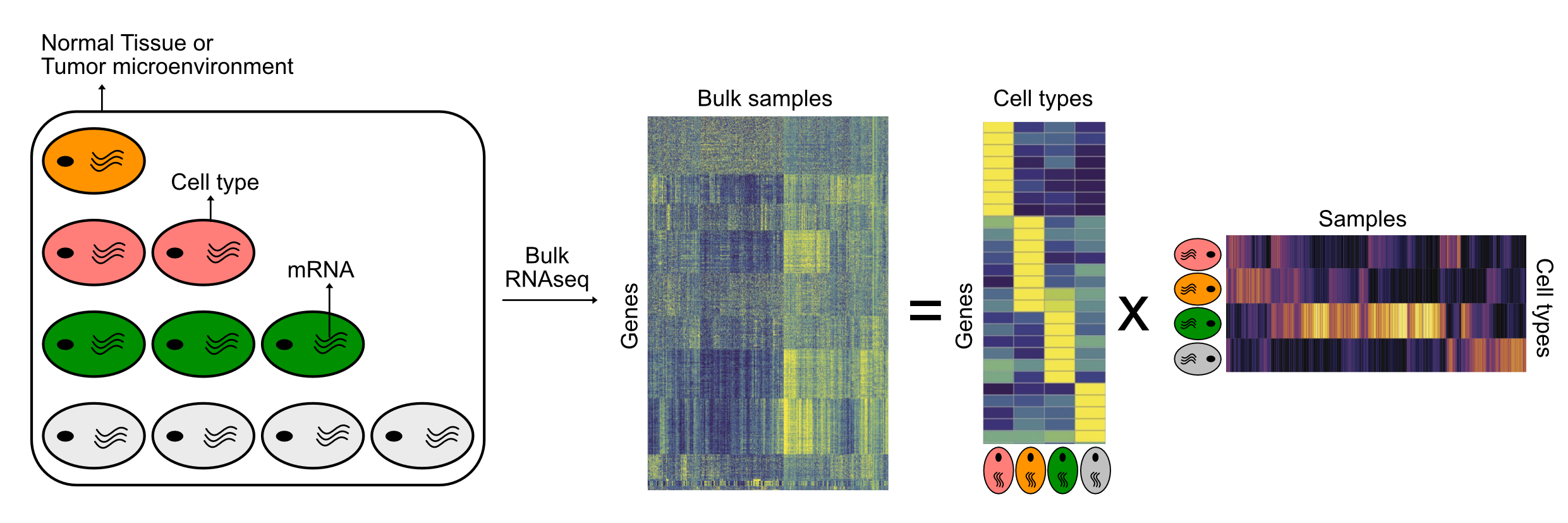
Schematic description of cell type deconvolution from mixed tissue samples.
Computational biology toolbox
Over the years, I have curated a bunch of reusable code-chunks for common computational biology tasks. These are essential tasks repeated across multiple projects and can be repurposed simply by “copy-paste-use” method with context specific modifications. Some of these tasks include - bulk RNAseq differential gene expression, single cell RNAseq analysis, feature selection (elastic nets, random forests), survival analysis, clustering, enrichment analysis, curating gene sets for important biological processes, transcription factor regulome constructions, networks, heatmaps, common graphical visualisation (and many more).
Schematic description of common bioinformatics tasks with (epi-)genomic data
Redox signalling in yeast
In this project, we aim to identify all the genes which would lead to strong changes of the redox status in a yeast cell within its cytoplasm and the mitochondria. In order to do this, we use a chemical sensor developed in the lab of Tobias Dick which can measure changes to the redox state inside these intracellular compartments. We apply this sensor across 4746 single gene yeast mutants across 3 different nutrient conditions to measure the effect of each mutated gene along with the nutrient environment on redox and identify genes with promote/inhibit reduction/oxidation potential in the cell given a particular nutrient environment.
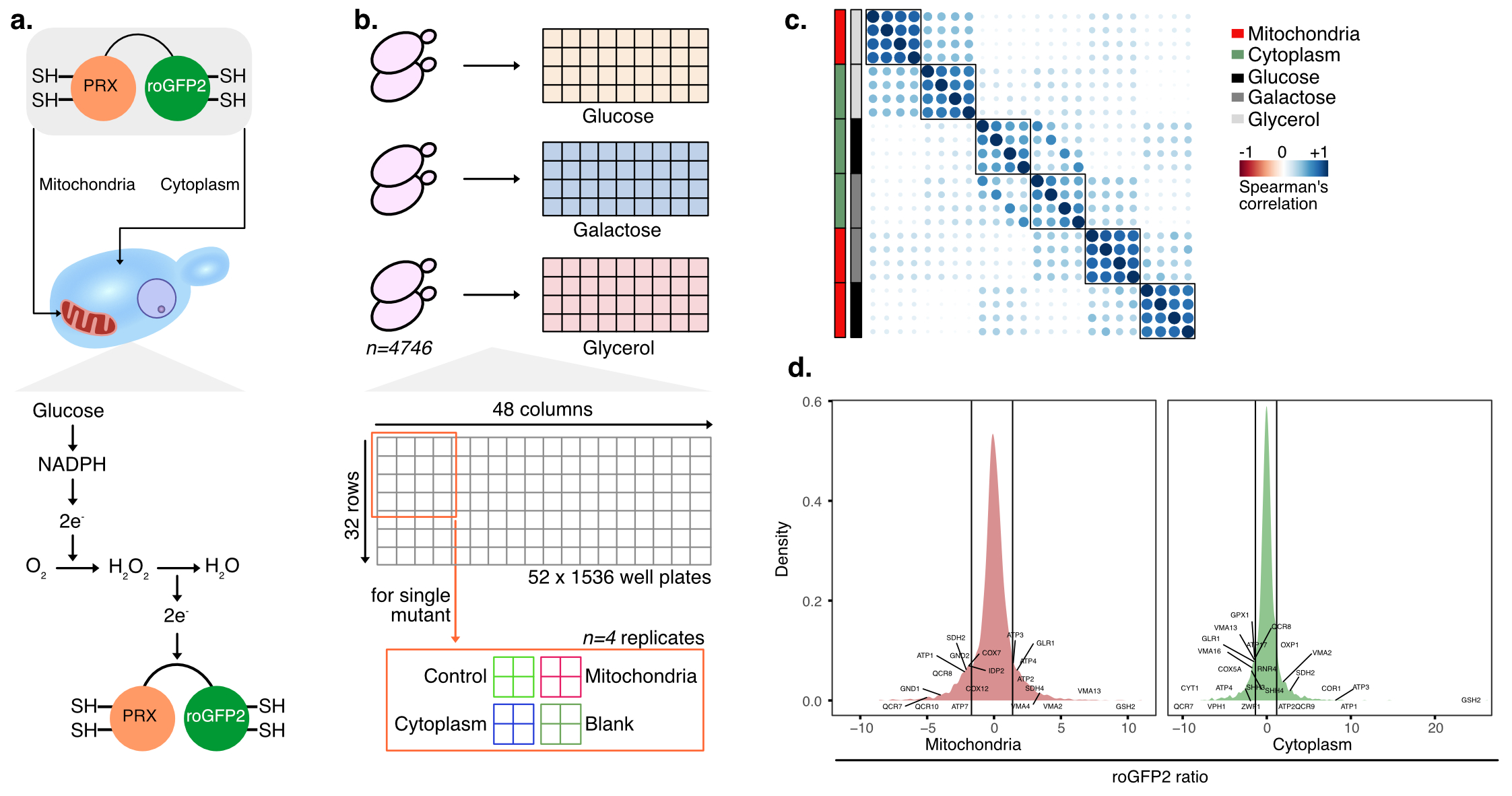
(a) Design of the chemical sensor to probe the redox status (b) Screening design - 4746 mutants were grown in 3 different nutrient conditions in 4 replicates to measure redox in the cytoplasm and mitochondria (c) Measuring screening quality by correlating the replicates (d) Identifying top hits (mutants) affecting reduction/oxidation
Computational phenotyping
In normal or diseased tissues, an intricate process involving the crosstalk between signalling, transcription factors and cellular metabolism takes place. These processes, by no means the only, are nonetheless the central biological processes which taken together defines the phenotypic state. Transcriptomic profiles capture snapshots of these processes and applying appropriate methods, it is possible to quantitatively compute scores which reflect the activity of these processes with reasonable accuracy. From these scores, one could then infer the complex crosstalk which defines the phenotypic space using latent factor models. Furthermore, I argue that at the phenotypic space it should be easier to classify biological conditions, akin to seeing the “bigger picture”.
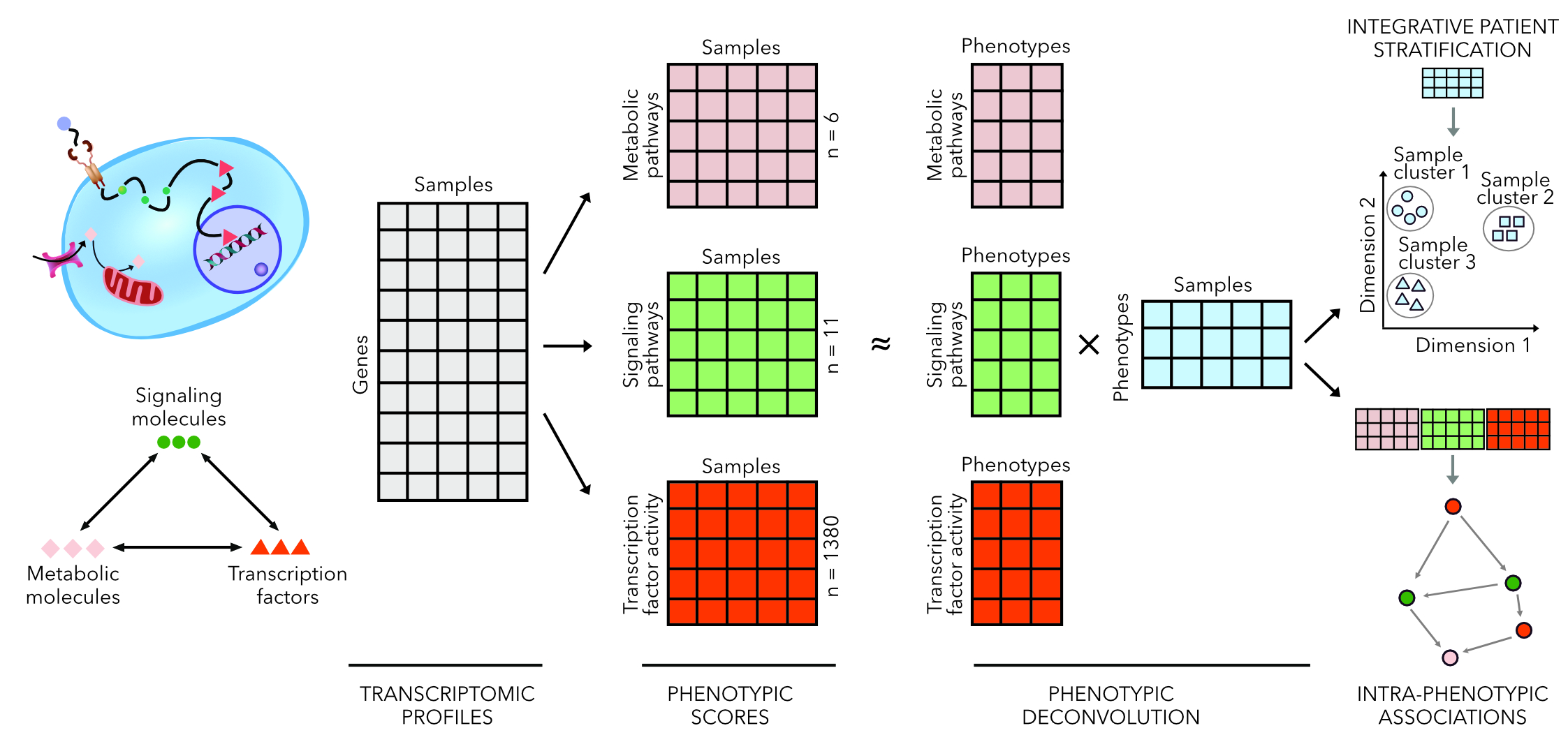
Proposed methodology for computational phenotyping
Other collaboration projects
I have also provided extensive bioinformatics support to a wide range of cancer biology research questions. For instance, in collaboration with Kirti Shukla we have shown that miR-30c-2-3p regulates NF-kappaB signalling and cell cycle by directly targeting TRADD and CCNE1 respectively in breast cancer and that this process is differentially modulated in ER- breast cancer processes.
In another project we explore the effect of Erufosine, an anti-cancer compound in oral squamous cell carcinoma cells and establish the mechanisms by which it inhibited tumor growth. In collaboration with Shariq Ansari we show that the inhibitory action of Erufosine is mediated by suppressing cell cycle and inducing endoplasmic reticulum and mitochondrial stress..
Next, in collaboration with Nisit Khandelwal, I was also involved in a project where he screened for immune checkpoint molecules that contributed to tumor resistance by inhibiting cytotoxic T lymphocytes. One of the top resistance causing hits was CCR9. Among others, I was specifically involved in analysis of the transcriptomic data generated form co-cultures of T cells and cancer cells having or lacking CCR9.
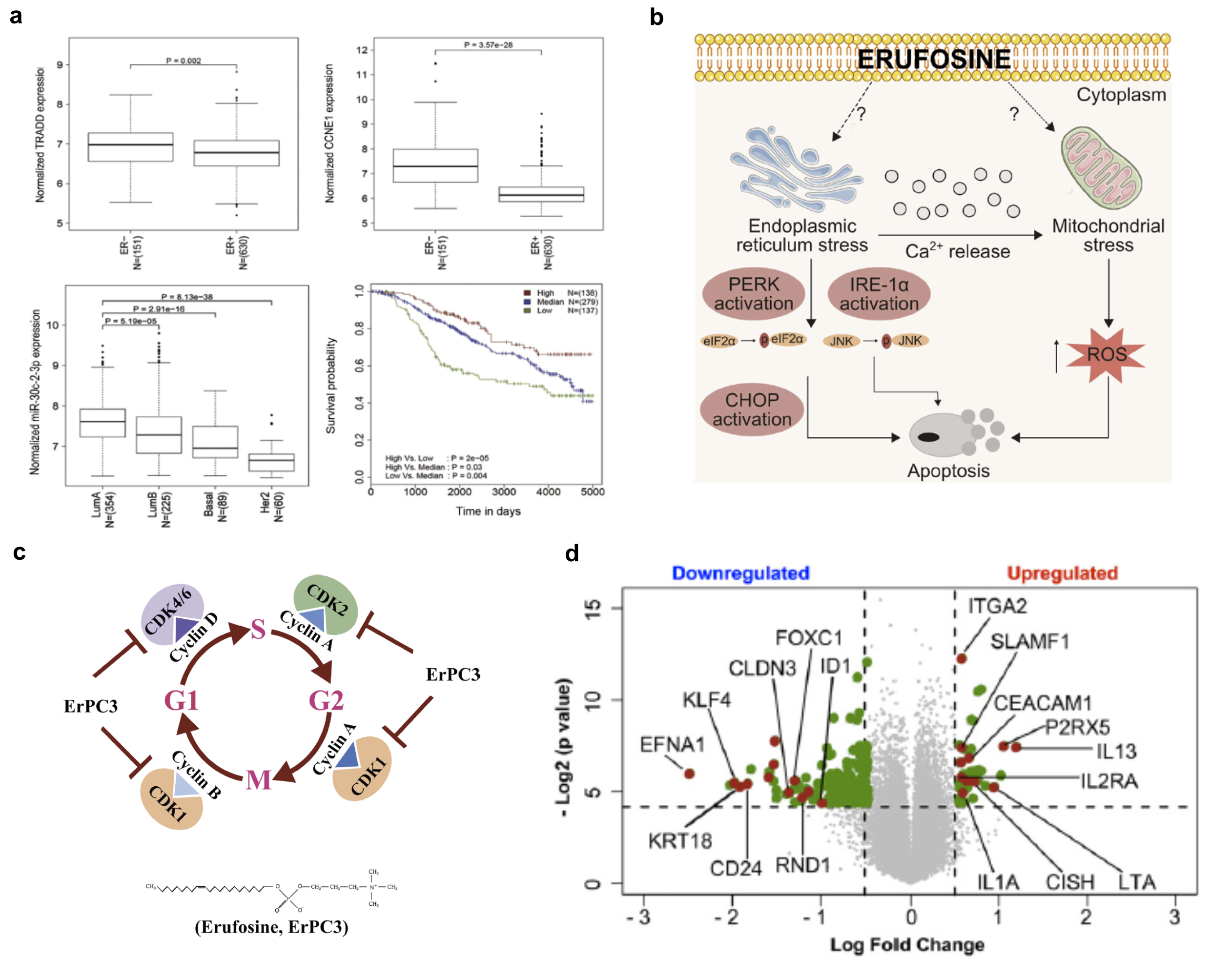
(a) Differential expression of miR-30c-2-3p and its targets TRADD and CCNE1 in ER- breast cancer (b,c) Mechanism of Erufosine anti tumor effect (d) Differential gene expression analysis of CCR9 high and low cancer cell (MCF7) grown in co-culture with Tcells